xCELLigence system continuously detects nerve cell culture Sebastian Diemert, Julia Grohm, Svenja, Tobaben, Amalia Dolga, Carsten Culmsee Abstract: To study the neuronal cell HT-22 and the response of primary cultured rat cortical neurons to different cell death stimuli, we used a real-time cell analyzer developed by Roche and ACEA Biosciences (Real-Time). Cell Analyzers, RTCA) The xCELLigence system uses non-invasive, label-free technology to continuously monitor neuronal cell HT-22. Studies have shown that by monitoring cell morphological changes, cell adhesion and cell proliferation, the xCELLigence system continuously monitors nerve cell culture and tracks changes in cells and their death. Keywords: nerve cell death, xCELLigence system, primary cortical neuron cells, HT-22 cells, cell impedance, real-time determination Foreword Neuronal apoptosis and necrosis are found in many neurodegenerative diseases. In recent years, several cell culture models have been established in neuroscience research for the study of cell death mechanisms in vitro. Although the scientific community has achieved certain results in the signal transmission principle of neuronal cell death in recent years, the technical limitations still exist, which limits the collection and interpretation of data. The inability to perform real-time monitoring of neuronal cell death is a major technical bottleneck. So far, the main methods of cell proliferation, cell survival and cell death detection are invasive endpoint detection, which is usually toxic to cells and requires destruction of cells. 1 Materials and methods 3 results Figure 1: HT-22 cell concentration gradient in E-Plate 96, and glutamate toxicity assay. (A) HT-22 cells of the indicated cell density were seeded on E-Plate 96 and recorded for 48 hours using the xCELLigence system. The cells were treated with glutamate 24 hours after inoculation. (B) HT-22 cell index (CI) values, normalized at the time point of glutamate addition. (C) After continuous xCELLigence cell monitoring, the MTT endpoint was performed in E-Plate 96 to test for cell viability. For the induction of HT-22 cell death, different doses of glutamate (3 and 5 mM) were used. In these cells, glutamate can cause the consumption of glutamine, thereby promoting the production of reactive oxygen species, leading to mitochondrial damage and cell death [2, 3]. No change in CI values ​​was detected during 8-10 hours after glutamate treatment. During the next 4-6 hours, the CI value decreases rapidly. The impedance plot reflects dose-dependent glutamate-induced cell death. The MTT experimental results of the E-Plate 96 reaction plate after impedance measurement further confirmed the results of this study (see Figure 1C). The cell death time was slightly different at different cell seeding densities and different glutamate concentrations. As shown in Fig. 1B, the xCELLigence recorded impedance map normalizes the CI value at a given time point of the drug. The glutamate-induced cell death dynamics detected by the xCELLigence system have good reproducibility over the time period and characteristics of glutamate-induced HT-22 cell death in previous studies, including mitochondrial rupture and nuclear AIF translocation [2, 3 , 4]. 3.2 CELLigence System Neuroprotection Assay To determine whether the xCELLigence system can detect neuroprotection, we used a small molecule inhibitor of pre-apoptotic BH-3 protein BID, BI-6C9, to prevent glutamate on HT-22 cells. Toxic effect. As shown in Fig. 2A, BI-6C9 was effective in preventing glutamate-induced apoptosis of HT-22 cells in the case where HT-22 cells were previously supplemented with 3 mM or 5 mM glutamate. The CI-6C9-treated cells recorded by the instrument, with or without the addition of glutamate, continued to increase in CI values, indicating that BI-6C9 maintains cell morphology and prolongs cell survival. Furthermore, cell proliferation (MTT) experiments were performed using E-Plate 96-well plates after termination of the experiment and additional standard 96-well plates set in parallel during the experiment, all demonstrating BI-6C9-mediated protection (see Figure 2B). ). These results further confirm that the xCELLigence system can be used to monitor cellular activity. The effect of the drug on the morphology of HT-22 cells was recorded by light microscopy (see Figure 2C). After the addition of glutamic acid, the cells undergo significant morphological changes, the cells are condensed and detached from the culture plate, and the impedance is lowered. This was further confirmed by the corresponding CI value monitoring records of the xCELLigence system, which showed that the CI value decreased after glutamate treatment. However, after the addition of BI-6C9, the morphology of HT-22 cells was unchanged, and the CI value record further confirmed the role of neuroprotection (see Figure 2A). Figure 2: Cell impedance measurement for neuroprotection of HT-22 cells. (A) The neuroprotective effect of the xCELLigence system on the detection of the Bid inhibitor BI-6C9. After inoculation, HT-22 cells were treated with BI-6C9 and/or glutamate and 24 hour cell index (CI) values ​​were recorded. (B) Cell viability was detected by MTT. The data in the left column is the detection after termination of CI by E-Plate 96, and the data in the right column is the detection in a standard 96-well cell culture plate. (C) The effect of the drug on the morphology of HT-22 cells was recorded by light microscopy. 3.3 Primary cortical neurons were cultured to isolate primary rat cortical neurons (PCNs) and plated on PEI-coated E-Plate 96-well plates at different cell densities per well. The inoculation density ranged from 8,000 to 32,000 cells/well. As shown in Figure 3A, the PEI coating does not interfere with the determination of cell impedance. The cells were cultured for 72 hours in a basal medium supplemented with 2% B27, and the primary neuronal cells were well adapted to the conditions of cell culture. Continuous monitoring of the xCELLigence system indicated that it was possible that within the first few hours of culture, the cells adhered to the bottom of the culture well and the CI value increased in the early stage of culture. In the different cell density wells, the display of the neural network was very clear by staining with PCNs microtubule-associated protein 2 (MAP-2) (see Figure 3B). To remove proliferating glial cells from primary cultured cortical neuronal cells, cultured cells were treated with 1 μM CAF for 3 hours on day 3 of culture. As shown in Figure 3B, treatment with CAF- resulted in a slight decrease in CI values, most likely a reflection of the removal of proliferating glial cells (see Figure 3B). Figure 3: Cell concentration gradient of primary cultured cortical neuronal cells. (A) Primary cortical neurons (PCNs) cells at specific cell densities were cultured and recorded for 6 days using the xCELLigence system. After primary isolation, PCNs were acclimated to the cell culture E-Plate 96-well plate environment for 3 days. Treatment with CAF for 48 hours to remove proliferating glial cells. Then continue to culture for 24 hours to facilitate cell recovery. (B) xCELLigence records show the effect of CAF-treatment on PCN cells. Cellular effects were normalized at the time point of CAF addition. (C) Staining pictures of MAP-2 in neuronal cells at different cell densities. To monitor the death of primary cultured neuronal cells (16,000 cells/well), cells were treated with calcium ionophore or glutamate. Cells were treated with calcium ionophores 155 hours after cell inoculation and found to have a significant decrease in CI values ​​5 to 6 hours after addition (see Figure 4A). The appearance of dense bodies was observed in primary cultured cells exposed to calcium ionophore compared to the control spindle-shaped flat bodies (see Figure 4). As shown in Figure 4B, after glutamate treatment, the CI value continued to decrease between 48 hours and 72 hours. Although glutamate is rapidly activated by the activation of the NMDA receptor, the intracellular calcium concentration is rapidly increased, but the subsequent cell death time is significantly delayed compared to the calcium ionophore-treated cells (compare pictures 4A and 4B). The reason may be that glutamate causes a slight delay in activation of cell death signals compared to rapid loss of cell membrane integrity and cell necrosis due to calcium ionophore treatment. Consistent with the above findings, optical microscopy showed that morphological changes in glutamate-treated cultured neurons were less pronounced than those treated with calcium ionophore (see Figure 4C), which is a dynamic record of the xCELLigence system. The results are consistent. Figure 4: Primary cortical neuronal cells were tested for cell death. (A) Cortical neuronal cells were treated with calcium ionophore on day 6 of culture and continued for 3 days using the xCELLigence system. (B) On day 6 of culture, growth factors were removed, primary cortical neuronal cells were treated with glutamate, and monitoring continued for 6-8 days. (C) The effects of drugs on primary cortical neuronal cells were recorded by observation with an optical microscope. 4 Conclusions Cellular degeneration and neuronal cell death are often associated with acute and chronic degenerative dysfunction disorders such as stroke, Parkinson's disease and Alzheimer's disease. The study of the mechanism of potential cell death in nerve cells is a prerequisite for determining the causes and treatment of neurodegenerative diseases, and is also of interest to researchers. In this study, we examined whether a well-recognized neural cell culture model can be used in the xCELLigence system and whether in vitro studies of neurotoxicity and neuroprotection can be performed. English version of the paper click to download references Source: Sebastian Diemert, Julia Grohm, Svenja, Tobaben, Amalia Dolga, Carsten Culmsee (2010), “Real-Time Detection of Neuronal Cell Death by Impedance-Based Analysis using the xCELLigence Systemâ€, Roche Diagnostics GmbH Roche Applied Science Werk, Penzberg 82372, Germany Sunson's complex Feed Enzymes were developed based on feed raw material characteristics, animal's nutritional needs, digestion system, productive performance and animal's welfare. The products can either be used as feed additive or dietary supplement to release more nutrients, eliminate anti-nutritional factors from feed raw materials or to make up for insufficient native enzymes in animal. The use of these products unambiguously indicated much improved daily performance of cow, cattle, poultry, swine, and many other animals. Feed enzyme complex,Feed additive enzymes,Feed supplement Sunson Industry Group Co., Ltd , https://www.sunsonchinaenzymes.com
Institute of Pharmacology and Clinical Pharmacy, University of Marburg, Germany
At the same time, the dynamic analysis of neuronal cell death and potential mechanisms requires a large number of experimental operations, covering a continuous, multiple time points. There are several end-point assays that allow the detection of specific molecular events at specific time points of apoptosis, such as phosphatidylserine translocation to the outside of the cell membrane, mitochondrial dysfunction, caspase activation, and DNA fragmentation [1] ]. One of the drawbacks of these endpoint assays is that they are unable to determine the time point of experimental apoptosis. To solve this problem, researchers need to repeat the monitoring of cell death morphological changes.
Currently, using the Real-Time Cell Analyzers (RTCA) xCELLigence system developed by Roche and ACEA Biosciences, the cells can be continuously monitored using non-invasive, label-free technology. The xCELLigence system integrates the sensor microelectrode lattice into the bottom of the E-Plate 96 culture well and records the impedance of the cells grown on it. Impedance measurement records the Cell Index (CI) value. When the cell is attached to the electrode, when the cell morphology changes, the current loop resistance changes, and the resistance is related to the cell index. The xCELLigence system tracks cell changes and cell death by monitoring cell morphology, cell adhesion and cell proliferation.
In this study, we used the xCELLigence system to study the response of different neuronal cell HT-22, and primary cultured rat cortical neuronal cells to different cell death stimuli. A feature of primary cultured nerve cells is their dense dendritic network, which can remain on the tissue reaction plate after apoptosis induction. Regardless of whether the cells themselves have undergone apoptosis, apoptotic neurons can remain attached to the cell culture plate and exhibit a certain resistance. The effect of this property on the monitoring of primary neuron culture is also a hot topic in this study using RTCA instruments. Moreover, we have studied the ability of the xCELLigence system to monitor neuroprotection. For this purpose, we use the neuroprotective agent BI-6C9, a well-recognized BH-3 small molecule inhibitor that interacts with a domain death agonist (BID), which is Bcl-2. Pre-apoptotic members of the gene family [4, 5].
In summary, studies have shown that the xCELLigence system can continuously monitor nerve cell culture through various experiments, including inoculation plate, pre-culture, proliferation of glial cells, drug effects, and neurotoxicity and neuroprotection. Confirmation of function.
1.1 HT-22 neuronal cells were seeded with HT-22 cells at a specified density on 96-well E-reaction plate 96 (Roche) or conventional 96-well reaction plates (Greiner, Frickenhausen) and grown in DMEM medium (Karlsruhe, Germany) In Invitrogen, 10% heat-inactivated fetal bovine serum (FCS), 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM glutamic acid (PAA Laboratories, Germany) were added to the medium.
1.2 Primary cortical neuron culture As described above, primary cortical neuronal cells are derived from the fetal rat brain (E16-18) [4]. Briefly, the cortical tissue is isolated, and after gentle pancreatin digestion, the neurons are mechanically separated and the meninges are removed. Different density cells were seeded in polyethylenimine (PEI) pre-coated with 96-well E-Plates 96 (Roche) or standard 96-well plates (Greiner). The medium was a neuronal medium (Invitrogen) supplemented with 5 mM HEPES, 1.2 mM glutamic acid, 2% (v/v) B27 supplement (Invitrogen) and gentamicin (0.1 mg/ml). After 48 hours of culture, treatment with cytosine-arabinofuranoside (CAF) for 48 hours inhibited the growth of non-neuronal cells. Then, after 6-7 days of in vitro culture, the medium was completely replaced, and the neuronal cells were used in the following experiment. To monitor changes in CI values ​​in glutamate-treated neurons, 1 μM CAF must be added to the cells on day 0 of culture.
1.3 Cell proliferation assay according to the cell proliferation kit I (MTT) (Roche) instructions, through the colorimetric MTT (3-[4,5-dimethylarsen-2-yl]-2,5-bromothiazole blue four The azole is tested to detect neuronal cell activity. The absorbance of the 570 nm well was read by an automatic FLUOstar Optima reader (Offenburg, BMG Labtech, Germany) with a reference filtration wavelength of 630 nm.
1.4 Real-time cell analysis CI values ​​were determined using the xCELLigence RTCA MP instrument with RTCA software 1.2 and continuously monitored in the E-Plate 96. Background impedance measurements of HT-22 cell culture were performed in 100 μl DMEM with 10% FCS, and background impedance measurements of nerve cell culture were performed using 100 μl of neuronal basal medium containing 2% B27. PEI coating does not interfere with cell impedance measurements. In contrast, poly-D-lysine or poly-L-lysine coating significantly interfered with real-time monitoring of primary cortical neurons. Continuous monitoring of HT-22 cells for 2 days, continuous detection of primary cortical neuronal cells for 12-14 days.
1.5 Fluorescence and optical microscopy Cortical neuronal cells were cultured on a PEI-coated IbiTreat μ-Slide 8-well reaction plate (Munich, Ibidi, Germany) for 6 days. Then, the cultured cells were fixed in paraformaldehyde at +4 ° C phosphate buffer (PBS) (pH 7.4). Neuronal cells were permeabilized using 0.2% Triton X-100/PBS using 10% (v/v) standard goat serum (NGS) and 2% (v/v) bovine serum albumin (BSA) PBS. It was terminated by treatment at room temperature for 1 hour. Microtubule-associated protein 2 (MAP-2) (Cambridge Abcam, ab11267, HM-2, 1:600 ​​dilution) mouse singles at +4 °C The cloned antibodies were incubated together and incubated overnight. On day 2, cells were incubated with goat anti-mouse secondary antibody (Alexa Fluor® 488 goat anti-mouse IgG (H+L) (Invitrogen, 1:250 dilution). Collected using a DMI6000 B inverted microscope (Leica, Germany) Fluorescence imaging information, analysis by LAS AF software (Leica Application Suite, Advanced Fluorescence 2.2.0, Leica Microsystems, Mannheim, Germany) using an Axiovert 200 microscope equipped with a Lumenera Infinity 2 digital camera (Ottawa, Lumenera, Canada) (Jena, Germany) , Carl Zeiss ) Obtain imaging information from an optical microscope. 10x 2.5 NA objective lens (Jena, Carl Zeiss, Germany) for daylighting, imaging capture by phase difference. INFINITY ANALYZE software (Lumenera) Digital imaging recording and imaging analysis.
3.1 Neuronal HT-22 cell proliferation To evaluate the growth and proliferation of the neural cell line HT-22, the cells were at different densities (4500, 8000).
Each well was incubated with E-Plate 96 and monitored using an xCELLigence RTCA MP instrument for 48 hours. After the cells adhered to the surface of the culture well, the impedance increased significantly. In the later experiments, the growth of HT-22 cells was stable. The final growth curves have very similar slopes, with only the absolute CI values ​​for each well, and the CI values ​​vary with the respective seeding densities (see Figures 1A and B). 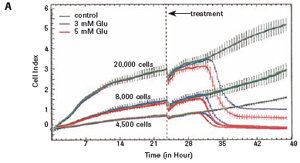
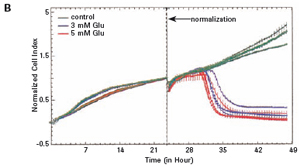
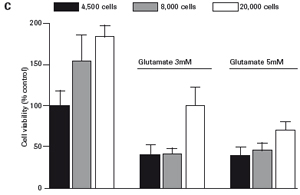


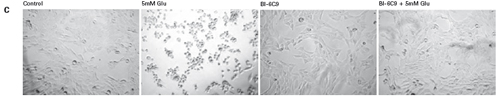
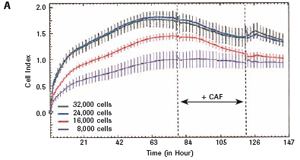
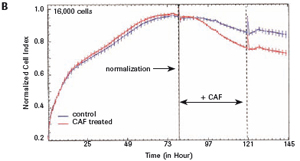
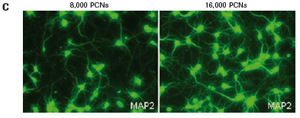
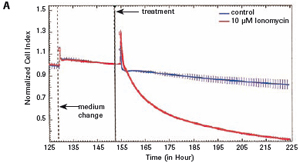

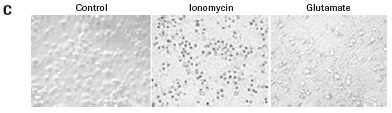
The study found that the xCELLigence system can be used to continuously monitor the neurotoxic effects of HT-22 cells and primary cortical neuronal cells. Treatment of HT-22 cells with glutamate rapidly initiates neuronal cell death and leads to its development, so it is traditionally difficult to determine the ideal time point for endpoint assays. xCELLigence displays neurotoxicity in real time through recorded impedance CI values ​​and pinpoints when downstream proteomics and genomic endpoints are detected. Moreover, the applicability of the xCELLigence system in neuroscience inhibitor research was further determined by experiments with the BID inhibitor BI-6C9 in neuroprotection. More importantly, the xCELLigence system allows us to monitor primary cortical neuronal cell culture conditions throughout the experiment, including plate inoculation, pre-culture, CAF treatment to remove glial cells, complex addition, and cell death maps.
In summary, through the xCELLigence system, we can monitor the cell culture conditions of the neural cell culture model, namely HT-22 cells and primary cortical neuronal cells. We found that this new method yields more information than traditional endpoint detection, while reducing the experiment itself and the time invested. Moreover, with the xCELLigence system, we can optimize the point in time for endpoint analysis and delve into the molecular mechanisms of neuronal cell death. Therefore, the xCELLigence system can be used as a very accurate and convenient tool for the study of neuronal cell death and neuroprotection in vitro.
[1]. Willingham MC. (1999). "Cytochemical Methods for the Detection of Apoptosis" J Histochem Cytochem 47(9): 1101-1110.
[2]. Landshamer S, Hoehn M, Barth N, Duvezin-Caubet S, Schwake G, Tobaben S, Kazhdan I, Becattini B, Zahler S, Vollmar A, Pellecchia M, Reichert A, Plesnila N, Wagner E, Culmsee C (2008). "Bid-induced release of AIF from mitochondria causes immediate neuronal cell death". Cell Death Differ 15(10): 1553-1563.
[3]. Grohm J, Plesnila N, Culmsee C. (2010). “Bid mediates fission, membrane permeabilization and peri-nuclear accumulation of mitochondria as a prereq¬uisite for oxidative neuronal cell deathâ€. Brain Behav Immun 2009 Dec 2 [ Epub].
[4]. Culmsee C , Zhu G, Landshamer S, Becattini B, Wagner E, Pellechia M, Blomgren K, Plesnila N. (2005). “Apoptosis-Inducing Factor Triggered by Poly (ADP-Ribose) Polymerase and Bid Mediates Neuronal Cell Death after Oxygen–Glucose Deprivation and Focal Cerebral Ischemiaâ€. Journal of Neuroscience 25 (44): 10262-10272.
[5]. Becattini B, Culmsee C, Leone M, Zhai D, Zhang X, Crowell KJ, Rega MF, Landshamer S, Reed JC, Plesnila N, Pellecchia M. (2006). “Structure–activity relationships by interligand NOE- Based design and synthesis of antiapoptotic compounds targeting Bid". Proc Natl Acad Sci USA. 103 (33): 12602-6.